RnaSec
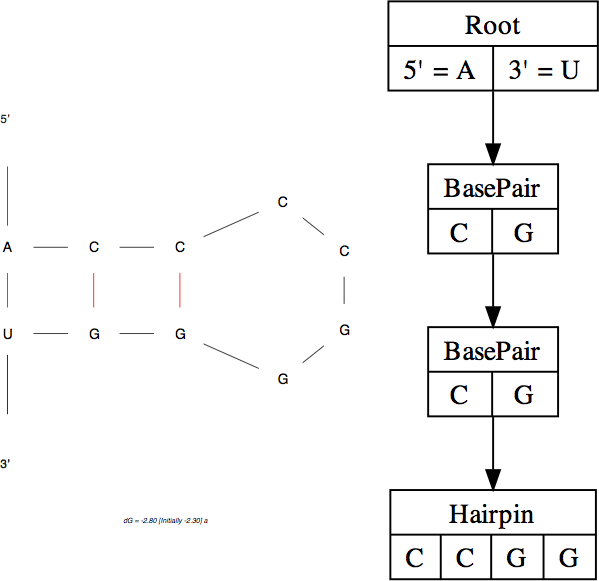
On the left, a folding generated by mfold shows two base pairs and a hairpin loop. This can be represented as a tree data structure shown on the right.

Screenshot of RnaSec’s unit tests running.
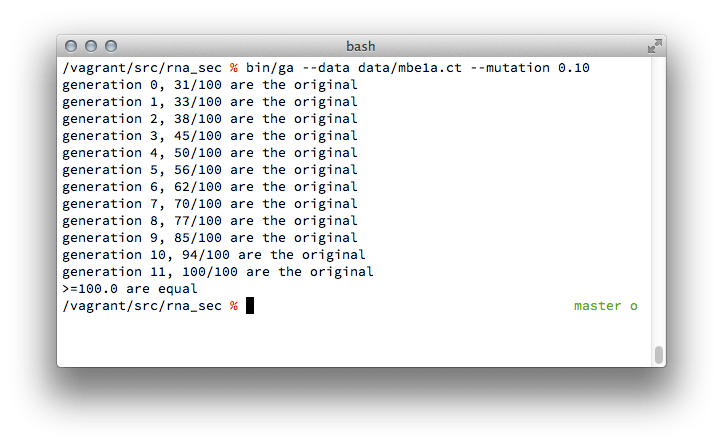
Screenshot of genetic algorithm running with 10% of the population available for mutation.
Background
In 2010, Doug Raiford (computer science) and Steve Lodmell (biochemistry) were collaborating in identifying RNA ligands that had high affinity for binding to the Rift Valley Fever Virus nucleocapsid protein. They had generated numerous aptamers for testing, but some were very similar (often just inverted), and since testing in vivo is expensive, they needed an in silico solution to determine which of these aptamers would be worth pursuing.
I developed RnaSec, a Ruby library for representing RNA secondary structures as tree data structures. It was a major stepping stone towards using tree-edit distance as a similarity metric between secondary structures.
What was the assignment?
This project spanned multiple courses:
- Introduction to Bioinformatics (Fall 2010): Represent RNA secondary structures as tree data structures (primarily parsing Vienna notation into objects).
- Machine Learning (Spring 2011): Write a genetic algorithm to permute a single secondary structure until the original structure is achieved. This was a proof-of-concept to test pruning and grafting operations.
- RNA Secondary Structure and Function Seminar (Spring 2011): Research using tree-edit distance and discuss the problem and approach with domain experts.
What am I most proud of?
I enrolled in the computer science program specifically to study bioinformatics, so becoming intimately familiar with a new domain (bioinformatics) and subdomain (RNA secondary structure) was eye-opening. While my interests shifted over my degree, I am most proud of learning to model a new domain, of being able to learn enough to speak competently with domain experts, and of developing this as a public-facing library with API documentation and unit tests.
What did I learn from the project?
I learned how to:
- quickly analyze and digest research papers and onerous dissertations
- write publishable libraries in Ruby
- write a genetic algorithm
- convey computer science-heavy topics to non-CS people (and have meaningful discussions as a result)
What would I do differently?
The goal of this project was to build a base foundation for creating a similarity metric between RNA secondary structures using tree-edit distance. While it delivered on providing an object-representation of RNA, it came at a great cost: much wasted time parsing undocumented plain text formats. Doing this over, I would modify one of the secondary structure predictors like mfold or UNAfold to label what it’s outputting. Tree-edit distance depends on identifying what a sequence is — whether it’s a bulge, hairpin, internal loop, loop, or base pair. However, the output from predictors is either an image or a mess of plain text. They already know structure they’re predicting, so it would only make sense if they output that!
Papers
- Report for Intro. to Bioinformatics
- Presentation for RNA Structure and Function Seminar
- Report for Machine Learning